Categoría: Health Sciences and Medicine
REVISIÓN
Mutations and laboratory diagnosis in acute promyelocytic leukemia
Mutaciones y diagnóstico de laboratorio en la leucemia promielocítica aguda
Elena Johanna Perez
Laborde1
![]() *,
Rosero Daniela Alexandra Freire1
*,
Rosero Daniela Alexandra Freire1
![]() *,
Marilin Castillo Yajaira Rueda2
*,
Marilin Castillo Yajaira Rueda2
![]() *,
Evelin Alexandra Zúñiga Sosa2
*,
Evelin Alexandra Zúñiga Sosa2
![]() *
*
1Universidad Técnica de Ambato. Facultad de Ciencias de la Salud, Carrera de Laboratorio Clínico. Ambato, Ecuador.
2Pontificia Universidad Católica del Ecuador. Sede Esmeraldas, Carrera de Laboratorio Clínico. Esmeraldas, Ecuador.
Citar como: Perez Laborde EJ, Rosero Freire DA, Rueda Castillo YM, Zúñiga Sosa EA. Mutations and laboratory diagnosis in acute promyelocytic leukemia. Salud, Ciencia y Tecnología - Serie de Conferencias. 2024; 3:554. https://doi.org/10.56294/sctconf2024554
Enviado: 09-07-2023 Revisado: 20-11-2023 Aceptado: 06-05-2024 Publicado: 07-05-2024
Editor:
Dr. William Castillo-González ![]()
ABSTRACT
Introduction: acute Promyelocytic Leukemia (APL) is a unique subtype of acute myeloid leukemia (AML) characterized by proliferation and accumulation of abnormal promyelocytes in the bone marrow. It arises from a balanced translocation between chromosomes 15 and 17, involving the retinoic acid receptor alpha (RARA) gene on chromosome 17 and the promyelocytic leukemia (PML) gene. It has a higher incidence in young adults.
Objective: to establish the mutations associated with acute promyelocytic leukemia and the techniques that aid in its clinical diagnosis.
Method: a systematic review of 19 scientific articles published in the databases of PubMed, Scopus, Google Scholar and the Virtual Library of the University of Granada was carried out. The data collected focused on acute promyelocytic leukemia, acquired, secondary or somatic mutations and laboratory diagnosis.
Results: of a total of 1730 patients 67,7 % had the t(15;17) mutation, of 682 patients 26,7 % had the FLT3-ITD mutation and of 175 patients 16 % had the FLT3-D835 mutation. Laboratory diagnosis is based on morphological evaluation of promyelocytes, hemostasis tests, biochemical tests Immunophenotyping and molecular cytogenetics.
Conclusions: the mutation associated with APL is the promyelocytic leukemia/retinoic acid receptor alpha (PML-RARA) gene, the same that presents a t(15;17), secondary mutations or somatic mutations such as (FLT3-ITD or FLT3-D835) were also known. Multiparametric flow cytometry is one of the most widely used techniques for the diagnosis of APL, allowing the determination of cell morphology and immunophenotypes.
Keywords: Acute Promyelocytic Leukemia; Acute Myelocytic Leukemia M3; Laboratory Diagnosis; Mutation.
RESUMEN
Introducción: la Leucemia Promielocítica Aguda (LPA) es un subtipo único de la leucemia mieloide aguda (LMA) caracterizada por proliferación y acumulación de promielocitos anormales en la médula ósea. Surge de una translocación equilibrada entre los cromosomas 15 y 17, que implica el gen del receptor alfa del ácido retinoico (RARA) en el cromosoma 17 y el gen de la leucemia promielocítica (PML). Tiene una mayor incidencia en adultos jóvenes.
Objetivo: establecer las mutaciones relacionadas con la leucemia promielocítica aguda y las técnicas que ayudan a su diagnóstico clínico.
Metodo: se realizó una revisión sistemática de 19 artículos científicos publicados en las bases de datos en PubMed, Scopus, Google académico y en la Biblioteca Virtual de la Universidad de Granada. Los datos
recopilados se centraron en la leucemia promielocitica aguda, mutaciones
adquiridas, secundarias o somáticas y diagnóstico de laboratorio.
Resultados: de un total de 1730 pacientes el 67,7 % presentaban la mutación t(15;17), de 682 pacientes el 26,7 % presentaba la mutación FLT3-ITD y de 175 pacientes el 16 % presentaban la mutación FLT3-D835. El diagnóstico de laboratorio se basa en la evaluación morfológica de promielocitos, pruebas de hemostasia, pruebas bioquímicas Inmunofenotipificación y Citogenética molecular.
Conclusiones: la mutación asociada a la LPA es el gen leucemia promielocítica/receptor del ácido retinoico alfa (PML-RARA), el mismo que presenta una t(15;17), también se conocieron mutaciones secundarias o mutaciones somáticas como (FLT3-ITD o FLT3-D835). La citometría de flujo multiparamétrica es una de las técnicas más utilizadas para el diagnóstico de LPA, permitiendo determinar la morfología celular e inmunofenotipos.
Palabras clave: Leucemia Promielocítica Aguda; Leucemia Mielocítica Aguda M3; Diagnóstico de Laboratorio; Mutación.
INTRODUCCIÓN
La Leucemia Promielocítica Aguda (LPA) es un subtipo único de la leucemia mieloide aguda (LMA) caracterizada por una proliferación y acumulación de promielocitos anormales en la médula ósea, provocando falla en la diferenciación, en el funcionamiento y deficiencia de la línea hematopoyética granulocítica.(1,2)
Según la clasificación Franco-Americana-Británica (FAB), la LPA morfológicamente posee dos subtipos, M3 hipergranular y la M3 variante hipogranular con características particulares en cada una de ellas.(3) Mientras que, según la última clasificación de la OMS del 2016, la LPA se clasifica dentro del subgrupo de la LMA caracterizado por anormalidades genéticas recurrentes, en particular, la leucemia promielocítica con la translocación.(4,5,6,7,8,9,10,11,12,13,14,15,16,17); PML-RARA.(4)
Epidemiológicamente en América Latina, mediante datos derivados del análisis realizado por el Consorcio Internacional de Leucemia Promielocítica (IC-APL), se estableció que esta enfermedad afecta principalmente a adultos jóvenes, con edades comprendidas entre los 18 y 40 años, sin distinción de género y la edad promedio de diagnóstico es a los 34 años. Además, se determinó que LPA tiene una baja incidencia en pacientes mayores a 60 años.(4)
Citogenéticamente la LPA se inicia con uno o más cambios adquiridos (mutaciones) en el ADN, las células de esta leucemia se caracterizan por la translocación recíproca muy específica que involucra al cromosoma 15 y 17 dando como resultado la fusión del gen leucemia promielocítica/receptor del ácido retinoico alfa (PML-RARA), la formación de este oncogén es el que conduce a la trasformación tumoral e interrumpe la diferenciación de las células leucémicas. El gen RARA (17q21.1) se fractura en el extremo 5’ y se fusiona con el extremo 3’ del gen PML (15q24.1) (figura 1).(2,3,5)

Figura 1. Esquemas que representan la formación de t (15;17). Translocaciones cromosómicas recíprocas (panel superior) y transcripciones de fusión (panel inferior)(6)
Las principales mutaciones somáticas en la LPA involucran especialmente al gen FLT3, que codifica la tirosina quinasa 3, son la duplicación en tándem interna (ITD) y una mutación en la región que codifica el ciclo de activación, en el codón del ácido aspártico 835 (D835).(5)
La fisiopatología de la LPA se debe a la interrupción en la diferenciación celular de los promielocitos,(1) mismos que viven de manera alargada debido a que la proteína de fusión PML/RARA ha demostrado prolongar la supervivencia de las células leucémicas al inhibir la apoptosis mediada por el factor de necrosis tumoral alfa. Los promielocitos experimentan una expansión clonal que resulta en una disminución de las células madre hematopoyéticas normales traduciéndose en anemia, neutropenia y trombocitopenia.(4)
Se ha visto que los promielocitos contienen gránulos cargados de proteínas y enzimas que al ser liberadas en el torrente sanguíneo causan la destrucción de los factores de coagulación, lo que unido al descenso de la cifra de plaquetas puede derivar en hemorragias muy graves.(1,4) Por otro lado, la LPA muestra un aumento significativo en la expresión de ciertas proteínas como la anexina A2 y el activador del plasminógeno tisular, lo que contribuye a un estado hiperfibrinolítico y aumenta el riesgo de coagulopatía por consumo y sangrado.(4,7) La proteína de fusión PML/RARA también activa directamente el factor tisular, lo que puede resultar en coagulación intravascular diseminada y aumentar el riesgo de trombosis.(8) Por lo expuesto, una particularidad de la LPA que la diferencia de las manifestaciones clínicas de otras LMAs y que aumenta su índice de mortalidad temprana son las coagulopatías graves, destacándose la coagulación intravascular diseminada (CID) y fibrosis sistémica.(9)
El tratamiento de primera línea de la LPA/M3 está dirigido con agentes diferenciadores de retinoides, como ácido transretinoico (ATRA), trióxido de arsénico (ATO), los mismos que disminuyen las complicaciones hemorrágicas causadas por la LPA. Incluso, una quimioterapia completa con ATRA ayudará a mejorar la supervivencia del paciente.

Figura 2. Acción del ATRA(10)
En la presente revisión bibliográfica, se pretende establecer las principales mutaciones relacionadas con la leucemia promielocítica aguda, mediante la identificación de genes asociados y las técnicas que ayudan a su diagnóstico clínico.
MÉTODO
Protocolo
El protocolo de la revisión sistemática fue realizado de acuerdo con la guía PRISMA (11)
Estrategia de búsqueda y criterios de elegibilidad
La búsqueda sistemática fue realizada en PubMed, Scopus, Google académico y en la Biblioteca Virtual de la Universidad de Granada. Las estrategias de búsqueda se personalizaron para cada base de datos (Anexo 1). Los estudios incluidos fueron revisiones sistemáticas, metaanálisis y estudios experimentales publicados en idioma inglés o español con una vigencia de cinco años o aquellos que aportaran información relevante sobre leucemia promielocitica aguda, también conocida como FAB M3, mutaciones adquiridas, mutaciones secundarias o mutaciones somáticas (FLT3-ITD o FLT3- D835) y resultados/eventos de interés como mortalidad, muerte temprana, supervivencia o pronóstico, herramientas diagnósticas/laboratorio. Se excluyeron estudios que evaluaban otro tipo de leucemia o que no informaban sobre mutaciones y otros tipos de estudios (comentarios, editoriales, cartas, informes de casos).
Selección de publicaciones y extracción de datos
La selección de las publicaciones fue realizada por los investigadores de forma independiente. Se preparó una tabla analítica y cada investigador decidió independientemente incluir o excluir una publicación después de evaluar el título y el resumen. Las discrepancias fueron resueltas en conjunto.
La información extraída de las publicaciones fue: autor(es), titulo, año de publicación, número de pacientes, WBC, datos de hemoglobina, mutaciones, inmunofenotipos y coagulopatias (anexo 2).
RESULTADOS
Resultados de la búsqueda
En la revisión sistemática se identificaron 19 artículos después de retirar publicaciones duplicadas. De estas, 8 fueron excluidas porque se consideraron irrelevantes en la búsqueda. La evaluación de los artículos completos fue realizada de 11 estudios, de los cuales seis fueron excluidos por no presentar las mutaciones de interés. Finalmente, un total de cinco artículos fueron incluidos en la revisión sistemática, como se muestra en la figura 3.

Figura 3. Diagrama de flujo para identificar los estudios sobre leucemia promielocítica aguda con mutaciones adquiridas en FLT3.
Todas las publicaciones reportaron datos de pacientes con LPA que
incluían un total de 2554 pacientes, con una mediana de edad de 51 años. De
estos pacientes 1730 (67,7 %) tenían el reordenamiento genético PML-RAR,
(15:17) (q22;q12) y 682 (26,7 %) la mutación FLT3-ITD. Solo una publicación
informó datos sobre la mutación FLT-D835 en 175 (16 %) pacientes de 1104.![]()
Cuatro publicaciones informaron sobre el riesgo elevado de tener un recuento de glóbulos blancos
≥10×109/L en pacientes con LPA asociados a las mutaciones de FLT3; tres publicaciones reportaron cifras bajas de hemoglobina con un valor medio de 93,8 g/L; y dos reportaron valores de recuento de plaquetas <50x109/L, este último asociado con el peligro de que el paciente pueda desencadenar una coagulación intravascular diseminada.
Los inmunofenotipos encontrados con alta intensidad en pacientes con LPA, en tres estudios, fueron todos marcadores mieloides (CD33, CD13, cyMPO) y en casos hipergranulares, los promielocitos expresaron solo CD117 un marcador blástico. Las expresiones fueron negativas para CD34.
El presente estudio tiene algunas limitaciones. La imposibilidad de recopilar mayor cantidad de publicaciones sobre los parámetros analizados y las diferentes formas de expresar el resultado, en algunos se incluyó el número total, mientras que en otros solo el porcentaje. Esto podría ocasionar sesgo en la revisión sistemática.
La tabla 1 presenta detalles del número de pacientes, datos de laboratorio y tipos de mutaciones extraídas de cada estudio:
|
Tabla 1. Características de los estudios incluidos en la revisión sistemática |
|||||||||||
|
Estudio |
N |
LPA |
Edad (años)* |
WBC (≥10×109/L) |
Hb (g/L) |
PLT (x109/L) |
t(15:17); PML- RAR % (n) |
FLT3- ITD % (n) |
FLT3- D835 n % (n) |
Fenotipo |
CID |
|
Kárai, 2019 (12) |
20 |
8 |
53 (32-74) |
17,01 (0,46-56,1) |
96 (77-115) |
38 (8-85) |
75 % (n = 6) |
37,5 % (n = 3) |
- |
CD117 (+), CD33 brillante, CD34 (-) MPO (+) |
100 % |
|
Matarraz, 2018 (13) |
109 |
109 |
54 (4-81) |
9,00 (0-127) 12 % (>30x109/L) |
100 (40-150) 94 % (Anemia) |
27 (2-190) 78 % (<50x109/L) |
100 % (n=109) |
31 % (n = 34) |
- |
CD33 brillante, CD34 (-) MPO (+) CD203c (+) |
89 % |
|
Chen, 2016 (14) |
327 |
51 |
50 |
- |
- |
- |
100 % (n = 51) |
- |
- |
CD33 (+), CD45 (+), MPO (+), CD13(+), CD117(+) |
- |
|
Pérez, 2019 (15) |
18 |
5 |
41 |
11,13 (1,76-28,72) 40 % (>10x109/L) |
85,4 (65-109) |
- |
60 % (n = 3) |
- |
- |
- |
80 % |
|
Picharski, 2019 (16) |
2381 |
2381 |
- |
15 % (>10x109/L) |
- |
- |
66 % (n = 1561) |
27 % (n = 645) |
16 % (n = 175) 1104 |
- |
- |
|
Total |
2855 |
2554 |
51 |
11,79 |
93,8 |
23,5 |
67,7 % (n = 1730) |
26,7 % (n = 682) |
16 % (n = 175) |
- |
90 % |
|
* Mediana |
|||||||||||
DIAGNÓSTICO DE LA LEUCEMIA PROMIELOCITICA AGUDA
Para el diagnóstico de la LPA se debe realizar una identificación de la morfología celular, citometría de flujo multiparamétrica (CFM), pruebas de coagulación, bioquímicas y la citogenética molecular que confirma el diagnóstico. (Tabla 2)
Morfología celular: se observan células atípicas (promielocitos) que presentan un núcleo bilobulado, el mismo que en la mayoría de las veces es oculto por la presencia de gránulos prominentes y bastones de Auer.
Pruebas de hemostasia: TP, TTPa, fibrinógeno y recuento plaquetario.
Pruebas Bioquímicas: glucemia, uremia, creatinina y ácido úrico.
Inmunofenotipo: Esto se lo realiza a través de la CFM proceso que permite la medida simultánea de diferentes características de una célula e inmunofenotípo que de manera inicial orientan al subtipo de leucemia mieloide aguda.
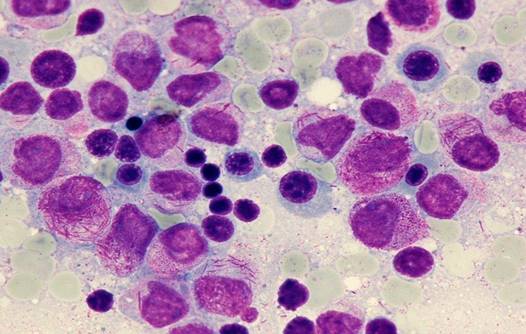
Figura 3. Promielocitos con presencia de bastones de Auer(17)

Figura 4. Técnicas utilizadas en el diagnóstico de Leucemia Promielocítica Aguda
Sin embargo, el diagnóstico definitivo de la LPA se basa en la detección del gen de fusión PML- RARA y la presencia de la translocación t(15;17) (q22;q21) que confirma el diagnóstico mediante la técnica de citogenética molecular. Existen diferentes técnicas para la identificación del gen PML- RARA entre estas el cariotipo convencional, hibridación fluorescente in situ o transcriptasa inversa y la reacción en cadena de la polimerasa. Sin embargo, las pruebas citogenéticas y moleculares son muy lentas en comparación a las pruebas de CFM que constituyen pruebas más rápidas para el diagnóstico de LPA.(14,15)
|
Tabla 2. Diagnóstico Diferencial(12) |
||
|
Pruebas |
LPA/M3 |
LPA/M3v |
|
Morfología |
Hipergranular Frecuentes bastones de Auer |
Hipogranular Bastones de Auer en proporción variable |
|
Leucocitos |
Bajos |
Hiperleucocitaria |
|
Inmunofenotipos |
CD34 -, CD33+, CD13 +, CD1 5 +, CD117, CD38 +. |
CD34 ±, CD33+, CD9+, CD13 +, CD117, CD38+. |
|
Plaquetas |
Trombocitopenia |
Trombocitopenia |
|
Hemoglobina |
Disminuida |
Aumentada |
|
TP |
Prolongado |
Prolongado |
|
TTPA |
Prolongado |
Prolongado |
|
Fibrinógeno |
Bajo |
Bajo |
|
Ácido úrico |
Elevado |
Elevado |
|
Urea |
Elevado |
Elevado |
|
Creatinina |
Elevado |
Elevado |
CONCLUSIONES
Las manifestaciones clínicas de una LPA se presentan con una rápida evolución, los signos y síntomas están relacionados con la coagulación intravascular diseminada (CID) y fibrosis sistémica, siendo estas las principales causas de muerte. El análisis estadístico señala que los pacientes con mutaciones FLT3-ITD tienen más probabilidades de tener recuentos de glóbulos blancos ≥10×109/L, presentar anemia y trombocitopenia en el momento del diagnóstico. Se realizó una revisión sistemática con un total de 21 artículos científicos en los que se identificó que la mutación asociada a la LPA es el gen leucemia promielocítica/receptor del ácido retinoico alfa (PML-RARA), el mismo que presenta una translocación recíproca que involucra al cromosoma 15 y 17, también se conocieron mutaciones secundarias o mutaciones somáticas como (FLT3-ITD o FLT3-D835). En la actualidad la citometría de flujo multiparamétrica (CFM) es una de las técnicas más utilizadas para el diagnóstico de LPA, permitiendo determinar la morfología celular e inmunofenotipos.
RECOMENDACIONES
Se recomienda ampliar la búsqueda de información para evitar sesgos en la revisión bibliográfica.
REFERENCIAS BIBLIOGRÁFICAS
1. Ng CH, Chng WJ. Recent advances in acute promyelocytic leukaemia. F1000Research. 2017;6(0).
2. Yilmaz M, Kantarjian H, Ravandi F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J [Internet]. 2021;11(6). Available from: http://dx.doi.org/10.1038/s41408-021-00514-3
3. Leukemia and Society Lymphoma. Información sobre la leucemia promielocítica aguda. WwwLlsOrg [Internet]. 2015;1–9. Available from: https://www.lls.org/sites/default/files/National/USA/Pdf/Publications/Spanish_APL_Fact Sheet 12_15.pdf
4. Mejía Buriticá L, Ocampo M, Torres Hernández JD, Ospina Ospina S, Vásquez Palacio G. Caracterización clínica y citogenética de pacientes con leucemia promielocítica aguda atendidos en un hospital universitario en la ciudad de Medellín, Colombia. Rev Colomb Hematol y Oncol. 2022;8(Suplemento 1):40–3.
5. Noguera NI, Catalano G, Banella C, Divona M, Faraoni I, Ottone T, et al. Acute Promyelocytic Leukemia : Update on the Mechanisms of Leukemogenesis , Resistance and on. Cancers (Basel). 2019;11(1591):1–21.
6. Pérez AC, Prada-Arismendy J, Castillo-Peñuela E, Castellanos W. Detección del gen fusión PML-RARA en pacientes colombianos con leucemia mieloide aguda. Ces Med. 2019;33(2):88– 99.
7. David S, Mathews V. Mechanisms and management of coagulopathy in acute promyelocytic leukemia. Thromb Res [Internet]. 2018;164(January):S82–8. Available from: https://doi.org/10.1016/j.thromres.2018.01.041
8. Mantha S, Tallman MS, Soff GA. What’s new in the pathogenesis of the coagulopathy in acute promyelocytic leukemia? Curr Opin Hematol. 2016;23(2):121–6.
9. Wang ZY, Chen Z. Acute promyelocytic leukemia: From highly fatal to highly curable. Blood. 2008;111(5):2505–15.
10. Shen PF. MOLECULAR MECHANISMS OF CHEMOPREVENTION AND THERAPY OF CANCER BY RETINOIDS. Vol. 9, Frontiers in Bioscience. 2663.
11. Urrútia G, Bonfill X. Declaración PRISMA: una propuesta para mejorar la publicación de revisiones sistemáticas y metaanálisis. Med Clin. 2010;135(11):507–11.
12. Kárai B, Habók M, Reményi G, Rejtő L, Ujfalusi A, Kappelmayer J, et al. A novel flow cytometric method for enhancing acute promyelocytic leukemia screening by multidimensional dot-plots. Ann Hematol. 2019;98(6):1413–20.
13. Matarraz S, Leoz P, Fernández C, Colado E, Chillón MC, Vidriales MB, et al. Basophil-lineage commitment in acute promyelocytic leukemia predicts for severe bleeding after starting therapy. Mod Pathol. 2018;31(8):1318–31.
14. Chen Z, Li Y, Tong Y, Gao Q, Mao X, Zhang W, et al. Stepwise discriminant function analysis for rapid identification of acute promyelocytic leukemia from acute myeloid leukemia with multiparameter flow cytometry. Int J Hematol. 2016;103(3):306–15.
15. Pérez AC, Prada-arismendy J. Detección del gen fusión PML-RARA en pacientes colombianos con leucemia mieloide aguda. CES Med. 2019;33(2):88–99.
16. Picharski GL, Andrade DP, Fabro ALMR, Lenzi L, Tonin FS, Ribeiro RC, et al. The impact of Flt3 gene mutations in acute promyelocytic leukemia: A meta-analysis. Cancers (Basel). 2019;11(9):1–14.
17. Montañés MR V. Promielocitos atípicos con astillas.
FINANCIACIÓN
Los autores no recibieron financiación para el desarrollo de la presente investigación.
CONFLICTO DE INTERESES
Los autores declaran que no existe conflicto de intereses.
CONTRIBUCIÓN DE AUTORÍA
Conceptualización: Elena Johanna Pérez Laborde, Rosero Freire Daniela Alexandra, Yajaira Marilin Rueda Castillo, Zúñiga Sosa Evelin Alexandra.
Investigación: Elena Johanna Pérez Laborde, Rosero Freire Daniela Alexandra, Yajaira Marilin Rueda Castillo, Zúñiga Sosa Evelin Alexandra.
Metodología: Elena Johanna Pérez Laborde, Rosero Freire Daniela Alexandra, Yajaira Marilin Rueda Castillo, Zúñiga Sosa Evelin Alexandra.
Redacción – borrador inicial: Elena Johanna Pérez Laborde, Rosero Freire Daniela Alexandra, Yajaira Marilin Rueda Castillo, Zúñiga Sosa Evelin Alexandra.
Redacción – revisión y edición: Elena Johanna Pérez Laborde, Rosero Freire Daniela Alexandra, Yajaira Marilin Rueda Castillo, Zúñiga Sosa Evelin Alexandra.